
Что такое ДЛКЛ?
Дефицит лизосомной кислой липазы (ДЛКЛ) — наследственное
заболевание, связанное с дефицитом фермента лизосомной кислой
липазы (ЛКЛ).
При недостаточности ЛКЛ нарушается процесс обмена жиров и
холестерина в клетках, что в свою очередь приводит к их
накоплению в клетках печени, селезенки, кровеносных сосудов,
слизистой тонкого кишечника, надпочечников и приводит к
различным сбоям в работе внутренних органов.1
Типы наследования
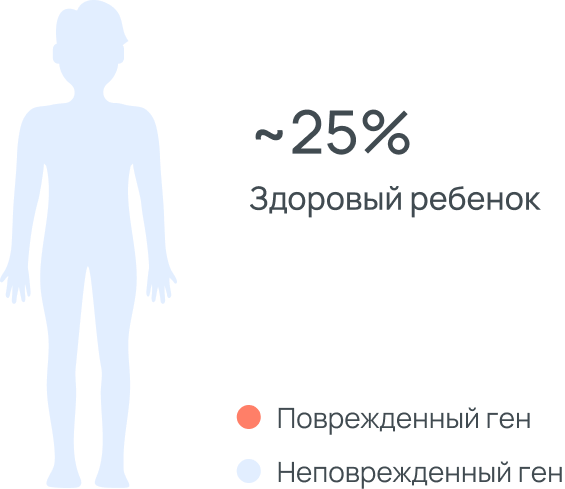
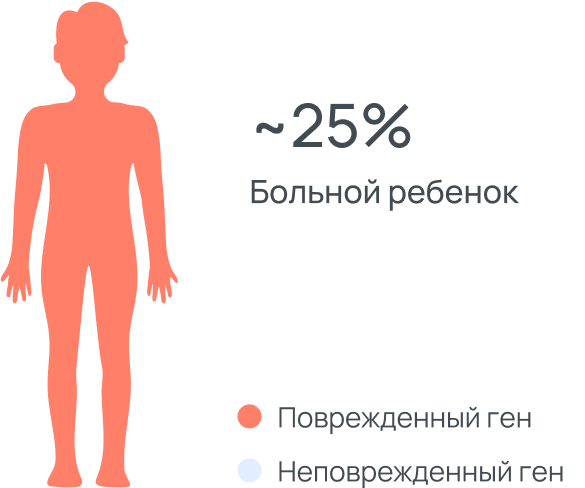
ДЛКЛ это аутосомно-рецессивное заболевание, это означает, что
оба родителя являются носителями мутации, но не болеют, т.к. у
них есть вторая копия здорового гена. Ребенок с ДЛКЛ наследует
одну копию «пораженного» гена от отца и одну от матери.
Заболевание проявляется только в случае наличия двух копий
пораженного гена. Риск рождения ребенка с ДЛКЛ в данном случае
составляет 25%. Носители болезни наследует только одну копию
пораженного гена либо от отца, либо от матери. Они не болеют и
никаких признаков болезни у них нет.

О ДЛКЛ за минуту
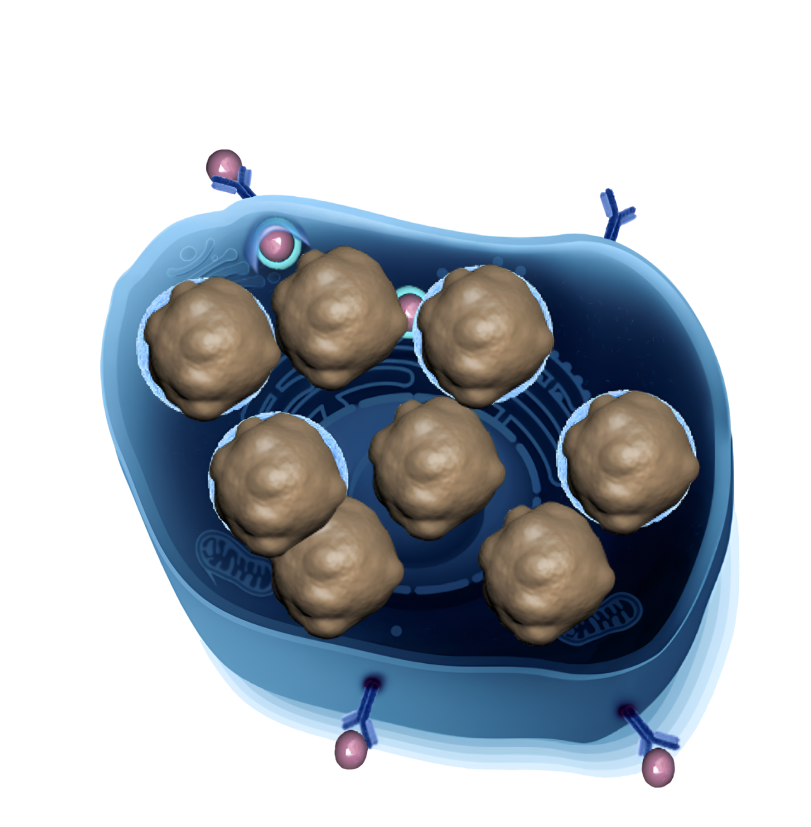
Роль ЛКЛ
Лизосомная кислая липаза — это фермент, который участвует в расщеплении жиров в клетках и находится в особых клеточных пузырьках - лизосомах.
В норме жиры расщепляются в лизосомах, а образующиеся жирные кислоты и холестерин усваиваются организмом для строительства новых клеток и для пополнения энергии.
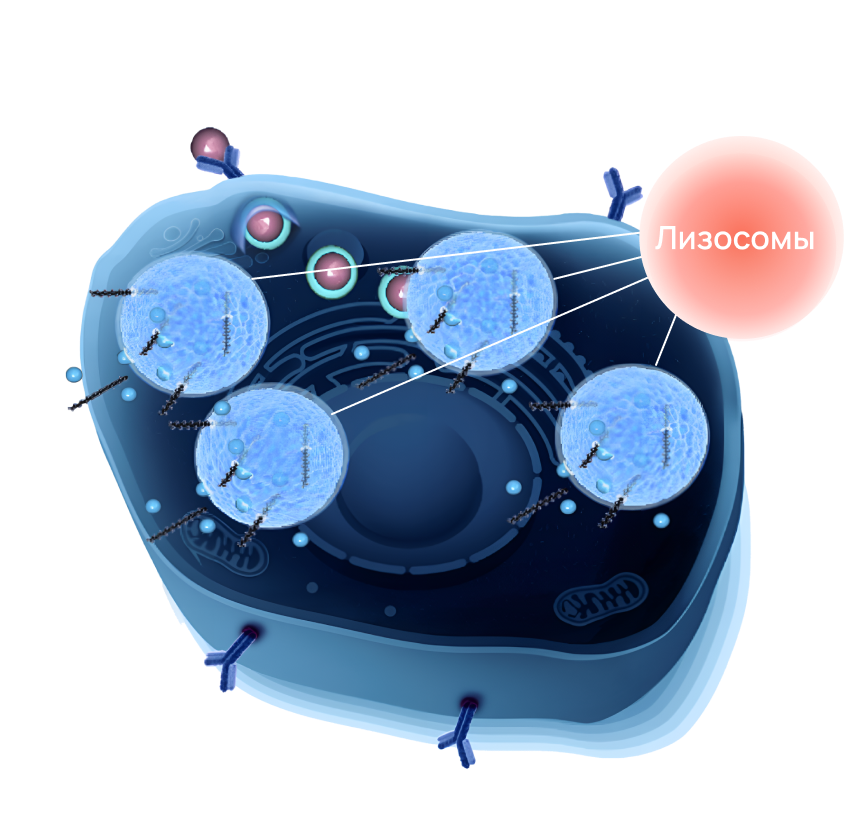
При ДЛКЛ фермент лизосомная кислая липаза (ЛКЛ) не работает должным образом. В результате лизосомы с неразрушенными молекулами накапливаются к клетках и нарушают их функции.
- Строкова Т.В., Багаева М.Э., Матинян И.А. Дефицит лизосомной кислой липазы. РМЖ. 2017; 19:1346-1351.
- Агеева Н. В., Агапова И. А., Амелина Е. Л. и др. Прогрессирующее заболевание печени: дефицит лизосомной кислой липазы (клинические наблюдения)//РМЖ. 2018. № 5(II). С. 96–103.
- Hoffman EP, Barr ML, Giovanni MA, et al. Lysosomal Acid Lipase Deficiency. 2015 Jul 30 [Updated 2016 Sep 1]. In: Adam MP, ArdingerHH, PagonRA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK305870/.
- Дегтярева А. В. и др. Болезнь Вольмана—тяжелая младенческая форма дефицита лизосомной кислой липазы//Неонатология: Новости. Мнения. Обучение.—2019.— Т. 7.—№ 2 (24).
- Jones A., ValayannopoulosV., Eckert S at all. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet Med.2016 May;18(5):452–8. doi: 10.1038/gim.2015.108.Epub2015 Aug 27.
- Burton BK, et al. Curr Med Res Opin. 2017;33(7):1211-1214.
- Русский медицинский журнал: https://www.rmj.ru/articles/pediatriya/Deficit_lizosomnoy_kisloy_lipazy/ (дата доступа: 02.12.2022).
- ЛПНП / ЛПВП / ГГТ - Липопротеины низкой плотности / Липопротеины высокой плотности / Гамма-глутамилтрансфераза.
- АЛТ / АСТ - Аланинаминотрансфераза / Аспартатаминотрансфераза.
- ЛДГ - Лактатдегидрогеназа.
- ДЛКЛ - Дефицит лизосомной кислой липазы
- ЛКЛ - Лизосомная кислая липаза